2007년
Next-Generation Sequencing(NGS) 기술이 개발된 이후 10여년간, NGS 기반 환자 코호트 연구를 통해, 다양한
mendelian disease와 암을 비롯한 여러 complex disease의 원인 유전자가 규명되어왔다. 하지만, PCR cluster
amplification 을 통해 신호를 증폭하여 염기서열 결정의 정확도를 높이는 NGS 기술의 특성상 시퀀싱이 진행될수록 에러 또한 증가하여,
시퀀싱 리드의 길이가 150bp~300bp 정도로 제한적인 문제가 있다. 최근 이러한 한계를 극복하기 위해, PCR 증폭(amplification)
없이 단일 DNA/RNA를 직접적으로 관찰함으로써 10Kb이상의 긴 DNA 조각을 한번에 시퀀싱할 수 있는 단일 분자 시퀀싱 (single-molecule sequencing), 혹은 롱-리드
시퀀싱(Long-read sequencing) 기술이 개발되었다. Read 길이가 증가함에 따라, 짧은 read 길이로는 발견하기 어려운 수천~수만개의
구조변이를 민감하게 관찰할 수 있게 되었고, 반복서열지역(repetitive region) 변이 분석 및 복잡한 구조변이의 재구성 또한 가능해졌다.
특히, Oxford Nanopore Technology(ONT)의 Nanopore sequencing 은 DNA/RNA 분자가 Nanopore
단백질을 통과할 때 발생하는 전류의 변화를 분석하는 원리의 염기서열 결정법으로, 최대 2Mb의 DNA 조각을 시퀀싱 할 수 있고, raw
gDNA 및 RNA를 시퀀싱할 경우 염기 변형(base modification) 확인을 통해 후성유전체(epigenome) 및 후성전사체(epitranscriptome)을
분석할 수 있는 장점이 있다.  그림 : Nik
Spencer/Nature ( Eisenstein, M. (2017). An ace in the hole for DNA sequencing. Nature, 550(7675), 285-288. doi:10.1038/550285a
)
본 프로젝트에서는, ONT의 high-throughput
sequencer인 PromethION을 활용하여 메틸화 (methylation) 정보가 남아있는 raw gDNA로부터 대장암 전장유전체(WGS)
데이터를 생산하고자 한다. Clinical sample의 특성상 짧은 DNA 조각이 많기 때문에, 이를 제거할 수 있도록 BluePippin을
통해 high-pass size selection(>8kb)이 수행된다. 이후 SQK-LSK109 ligation kit를 사용해
library prep이 진행되고, PromethION에서 72시간 이상 시퀀싱이 수행되며, 샘플당 60Gb ~ 100Gb 정도의 데이터가 생산된다.
이후 생성된 FASTQ는 adaptor trimming 및 basecall quality filtering을 거치고, long-read 전용
mapper(NGMLR 등) 을 통해 표준 유전체에 매핑된다. 생산된 Nanopore WGS 데이터는 대장암 특이적 구조변이 및 후성유전학적 변이
분석에 활용될 예정이다. Long read 기반의 somatic SV caller를 개발하여 대장암 특이적 구조변이를 확인하고, 이를 기반으로
전반적인 SV profiling을 진행할 계획이다. 또한, normal genome 상에도 구조변이가 다수 존재하기 때문에, patient
specific genome assembly를 구축함으로써 cancer
genome의 정확한 구조를 파악하고자 한다. 후성유전체 측면에서는, 5-hydroxymethyl -cytosine 분석을 통해 대장암 특이적
enhancer dysregulation 패턴을 파악하고자한다. 더 나아가, 유전체 및 후성유전체 데이터가 동일한 read에 존재하는 점을 활용하여,
methylation 으로 deconvolution이 가능한 cell subpopulation에 특이적으로 발생하는 구조변이를 파악 하고자한다.
이를 통해, 대장암 특이적 진단 및 예후 마커를 발굴할 수 있을 것이고, 대장암 생성 및 발달 메커니즘을 확인할 수 있을 것으로 기대된다. 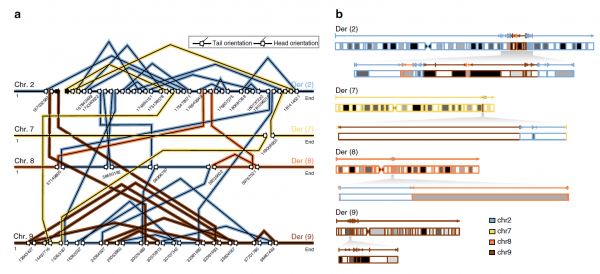
그림 : Nanopore
sequencing data를 이용해 재구성된 large chromothriptic events의 예시 ( from Nat Commun.
2017 Nov 6;8(1):1326 ) |
참고 문헌 - Eisenstein, M. (2017). An ace in the hole for DNA sequencing. Nature, 550(7675), 285-288. doi:10.1038/550285a
- Stancu, M. C., Roosmalen, M. J., Renkens, I., Nieboer, M. M., Middelkamp, S., Ligt, J. D., . . . Kloosterman, W. P. (2017). Mapping and phasing of structural variation in patient genomes using nanopore sequencing. Nature Communications, 8(1). doi:10.1038/s41467-017-01343-4
- Mantere, T., Kersten, S., & Hoischen, A. (2019). Long-Read Sequencing Emerging in Medical Genetics. Frontiers in Genetics, 10. doi:10.3389/fgene.2019.00426
|

