수많은 유전자
중 암과 관련된 유전자는 약 500 여개로 추정되고 있다. 이러한
연구 결과를 바탕으로 암 환자의 유전적인 변이를 확인하기 위해서 WGS (whole-genome
sequencing) 또는 WES (whole-exome sequencing)의 적용은
경제적으로 매우 비효율적이다. 이러한 한계를 극복하고자 특정 유전자만을 high-depth로 데이터를 생산할 수 있는 panel sequencing
(targeted-sequencing)이 존재하며, 이렇게 high-depth의 데이터는 임상적으로 유의미한 변이들을 더욱 정확하게 관찰 할 수 있다는 장점이 있다. DNA 내에 낮은 비율로 존재하는 암
특이적 변이를 확인하기 위해서는 high-depth 이외에도 실제 변이와 PCR bias 또는 Sequencing error의 구분이 필요하며, 변이의 정확한 구분은 이후 환자의 관리 및 치료에 적절한 방법을 제시할 수 있는 중요한 인자이다.
본 프로젝트에서는
우선 대장암 환자에서 고빈도로 발견되는 변이 유전자를 대중적으로 사용되는 암 관련 데이터베이스를 기반으로 선정하였으며, 선정된 유전자들의 모든 Exon 및 actionable genes에 대한 promoter/enhancer 영역
또한 표적에 포함시켜 총 46개의 유전자에 대한 대장암 특이적 패널을 제작하였다. (그림. 1 암
관련 유전자 변이에 관해 존재하는 다양한 데이터베이스) 이후에 라이브러리
제작 단계에서 UMI를 도입시켜 앞서 언급한 PCR bias 또는
Sequencing error를 정확하게 구분할 수 있는 기술을 적용하였다. UMI가 적용된 데이터는 자체적으로 개발 및 테스트된 분석 파이프라인을 기반으로 전처리가 진행되었으며, 이 과정에서 상당히 낮은 비율로 존재하는 변이를 발견할 수 있는 것을 확인할 수 있었다. 전체 리드(read) 중 우리가 원하는 표적 지역에 정렬이 되는
비율은 약 73%로 상당히 높은 것을 확인하였고, 표적 지역에
depth 또한 > 500 이상인 것을 확인할 수 있었다. 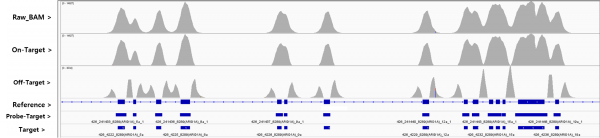
(그림. 2 IGV를
통해 관찰한 sequencing read의 분포)
이를 기반으로 동일 환자의 WGS 데이터와
표적 지역에서 발견된 변이 및 allele frequency (AF)를 비교해본 결과 panel-sequencing에서만 발견된 변이들의 AF는 상당히 낮은
비율로 존재하는 것을 확인할 수 있었다. 따라서 tumor heterogeneity
한계를 극복하기 위해 panel-sequencing의 도입은 적절하다고 판단할 수 있다. 이렇게 발견된 변이들은 각 종 데이터베이스를 기반으로 Tier를
메겨 임상적으로 얼마나 유의미한지 구분하였고, 이를 기반으로 리포팅 시스템 구축을 통해 임상의들의 진단
및 추후 환자 관리 효율을 극대화를 하는 것을 목표하고 있다. (그림. 3 데이터베이스 기반의 변이 Tier 선정 기준) | 


