대장암 환자의 유전체 변이를 탐색하기 위한 Whole Genome Sequencing 데이터를 가공을 목표로 한다. GATK의 Preprocessing 을 이용하여 데이터를 가공했다. Picard, BWA, gatk로 raw fastq 데이터를 mapping, markduplicate과 BQSR을 진행했다. 또한 fastQC, Qualimap, duplicate stats, NGScheckmate 등을 이용하여 데이터의 QC를 진행했다. 1. fastq to unmapped reads conversion (readgroup 정보) (picard) 2. markillumina adapter (picard) 3. map to reference (hg38) : (bwa-0.7.17 mem) 4. Merge bam files : (picard) 5. markduplicate : (picard) 6. BQSR : (gatk4.0.2.1) - KnownSites: Homo_sapiens_assembly38.known_indels.vcf, Mills_and_1000G_gold_standard.indels.hg38.vcf, resources_broad_hg38_v0_Homo_sapiens_assembly38.dbsnp138.vcf 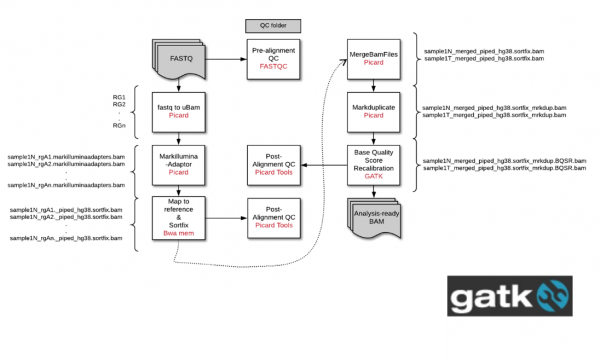
Variant Calling 은 Mutect2 와 Manta/Strelka2로 진행했다. 또한 variant 들의 annotation은 snpEFF, annovar과 vep 로 진행하여 데이터를 생산했다. 이와 같은 variant 데이터들은 Strelka2와 Mutect2의 merged set, intersected set으로 나누어서 분양을 진행되었다. 또한 somatic variant discovery refinement 를 통하여 tier1, 2, 3의 set를 생산했다.
| 

